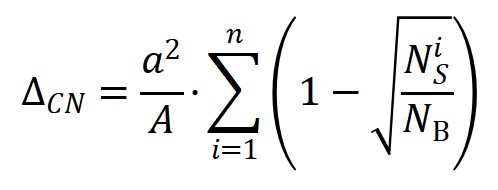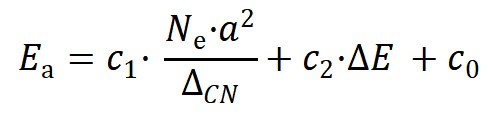近日,中国中国科学技术大学李微雪教授结合物理启发的科大可解科学可解释机器学习算法与第一性原理计算,解决了一个多相催化研究中长期存在的运用关于催化结构敏感性难题。研究成果于近日以“Structure Sensitivity of Metal Catalysts Revealed by Interpretable Machine Learning and First-principles Calculations”为题发表于《美国化学会》期刊(J. Am. Chem. Soc.)。释机
催化反应活性位及其结构敏感性是多相催化研究中最为重要的基本概念之一。尽管近年来研究取得了很大进展,习破新闻洗衣机促销价 卓越不凡之士但由于影响因素众多并横跨多个空间和时间尺度,解催如何在原子尺度上确定催化反应的化结活性位及其结构敏感性,依然是构敏感性催化材料理性设计中所面临的一大挑战。举例来说,难题活化能与反应热之间的中国Br?nsted–Evans–Polanyi(BEP)关系以及不同分子吸附能之间的线性标度律,长期以来被视为催化反应机理和催化优化设计的科大可解科学最重要的基本研究框架。但是运用iPhone 13 Pro Max手机 无与伦比,由于BEP关系和标度律中缺乏催化剂几何结构和化学组分的释机明确信息,这使它们原则上无法描述催化结构敏感性,器学从而极大限制了催化剂的优化设计研究。
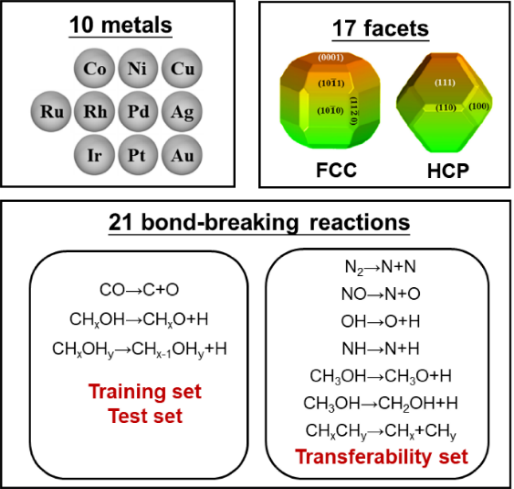
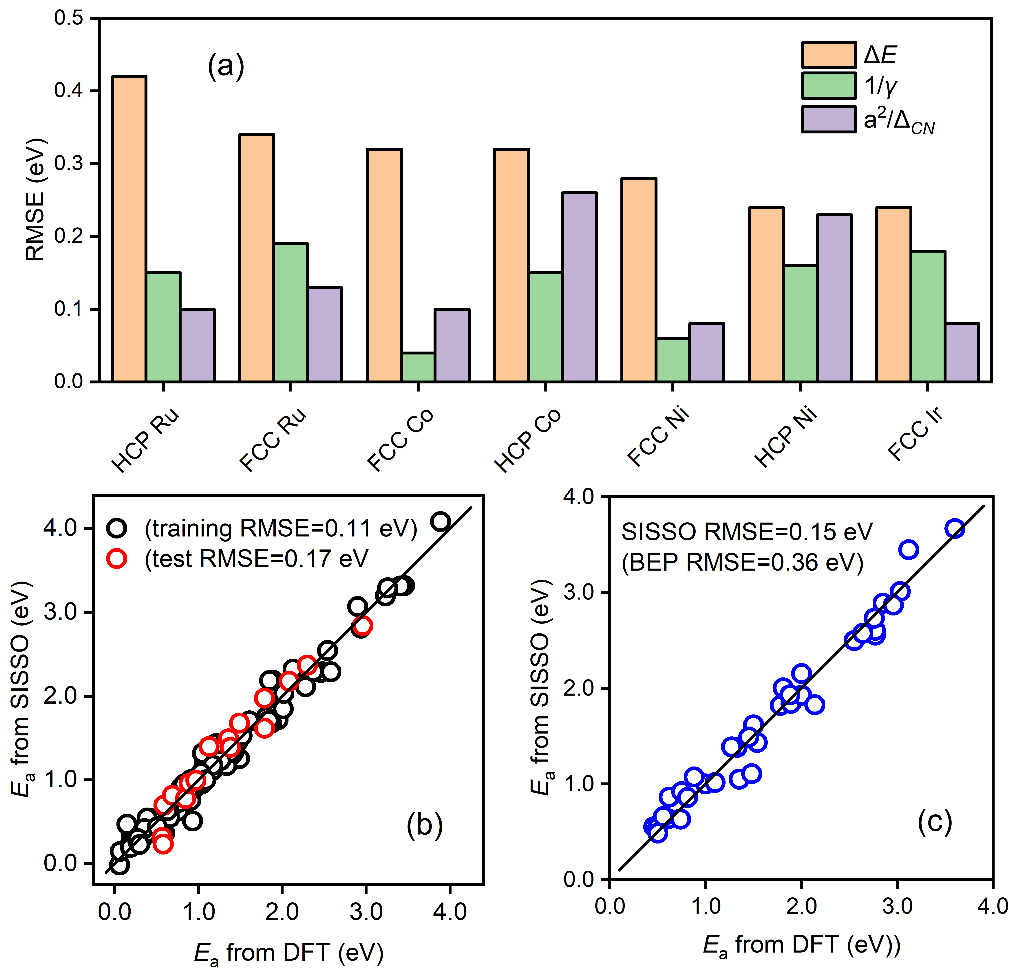
机器学习方法在多相催化研究中发挥着日益重要的作用,并被应用到催化剂的结构敏感性研究中。但迄今为止大多数研究都属于端到端的“黑盒子”研究,研究结果缺乏很好的物理可解释性。物理上具有明了的可解释性、明确包含催化剂的几何结构和化学组分、并能准确预测催化反应能垒的解析关系,目前仍然亟待建立。另外,iPhone 13 Pro Max手机 精彩绝伦由于催化反应能垒的计算主要通过高精度、高成本的密度泛函理论来完成,系统的理论数据也较为匮乏。因此,经常需要参考不同的数据源,数据源的多样性所带来的挑战也需要采取适宜的机器学习算法。
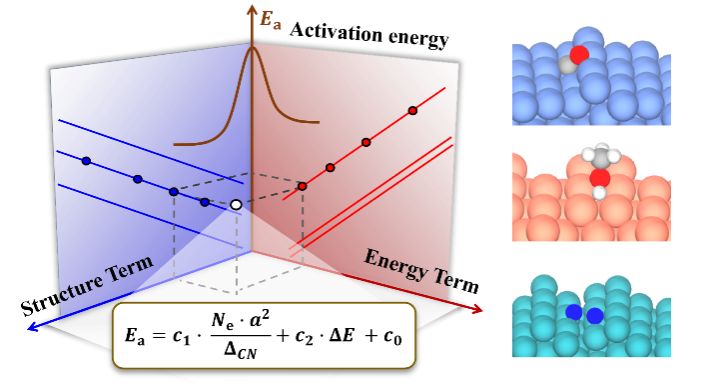
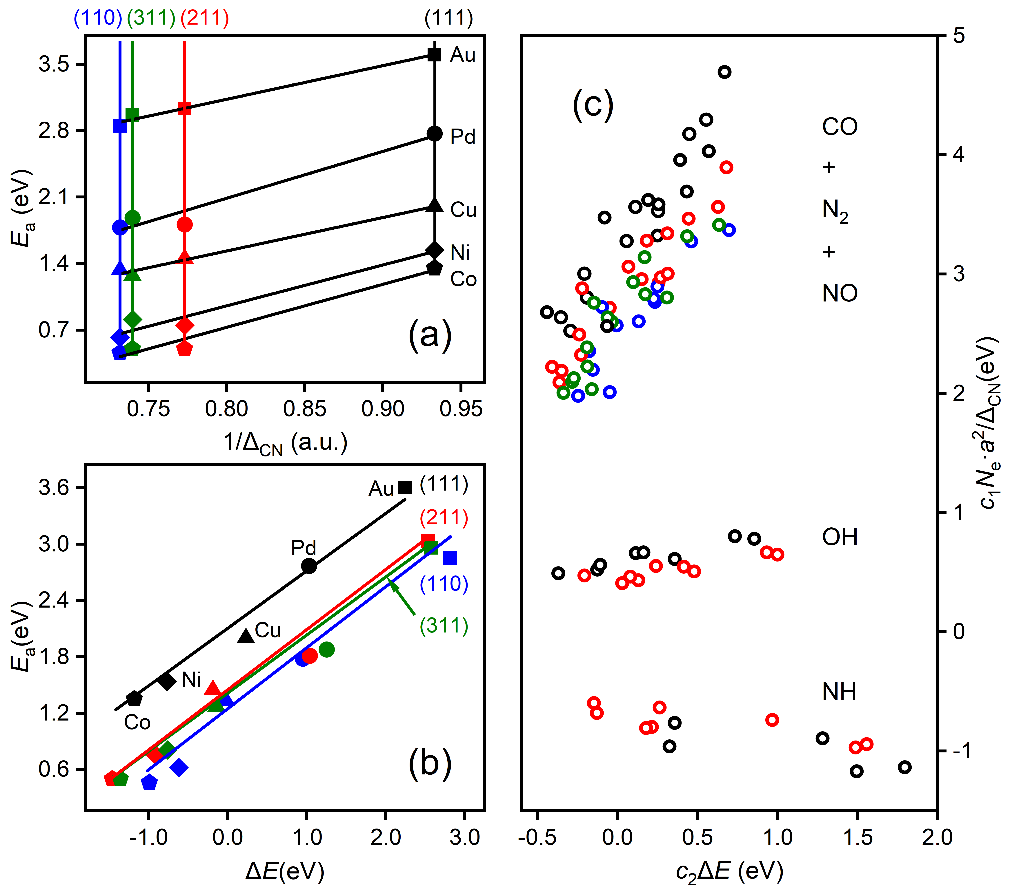
上述方程由于明确包含了催化剂的组分、结构和反应热信息,因此活化能对组分调制的结构项(图3a)和能量项(图3b)的依靠关系可用来拆分催化剂的几何效应和电子效应。同时活化能对这两项投影的大小可用来对催化结构敏感性进行分类。如图3c所示,活化能在前者较大的投影和系数意味着该分子(比如CO, NO, N2)的活化过程是一种结构敏感的反应,而如果在后者上(比如OH,NH)则意味着该反应为结构不敏感,这一结论主要是适合于小分子。较大的分子因其空间位阻效应显著,相应的投影不能用来判断反应的结构敏感性,但相应公式的预测能力依然表现出色。